Facilities & Equipment
101 ECM (Engineering Change Management)

Jun
As Stephen Hawking once said: “Intelligence is the ability to adapt to change” It’s a known fact that change is inevitable and constant. We deal with change daily in all aspects of our lives, including work, where we assess changes to understand how to deal with them in a way that will not impact the quality of the product we make. Yet, the pharmaceutical industry does not adopt or embrace change quickly.
In this blog posting, I will explain how the utilization of the RBA (Risk-based Approach) could be a very effective and time-saving tool when dealing with changes related to equipment and facilities.
Also, it will explain how to categorize these changes and implement them adequately without impacting the quality of the products or slowing down the process.
As per ISPE C&Q Baseline Guide Volume 5. 2nd edition 2019, the definition of change is as follows:
A system is considered to be changed when it is modified, altered, added to, removed, or improved in a way that makes its functions, physical features, or performance different from what they were before the change.
In the strictly regulated Pharma World, it is necessary to document and assess all changes to validated processes/equipment/facilities that may directly or indirectly impact product quality. Thorough review and assessment of any changes before their implementation ensure product quality and patient safety consistency.
To control changes that were determined to have a GMP or SISPQ (Safety, Identity, Strength, Purity, and Quality) product impact, many manufacturing companies utilize a change control process – could be paper-based or electronic application. However, QCC (Quality Change Control) program could become overwhelmed with unnecessary changes that do not impact the SISPQ of the products: and this is when RBA comes into play.
If too many non-GMP and non-SISPQ product impacting changes are added to the QCC system, it can result in some disadvantages like:
- Clutter the system with numerous unnecessary records.
- Heavily occupy personnel supporting quality change control process and reduce productivity.
The time spent to process a non-GMP/SISPQ-related change through QCC (including the implementation time) could lead to a cycle time of many weeks, if not months.
I believe that such practice (i.e., having non-GMP changes processed through quality change control) might show a lack of understanding of the change control program, especially if you explain it to auditors.
The non-GMP and non-SISPQ product impacting changes typically require minimum documentation and relatively simple execution/implementation; a risk-based approach to identify and detour such changes from the quality change control system can be implemented.
This solution aligns perfectly with the ISPE C&Q Baseline Guide Volume 5. 2nd edition 2019, which describes that the level of documentation of the change should be commensurate with the level of risk related to the change.
In other words:
- If the change is a low risk and simple, then the documentation should be minimal and straightforward.
- If the change is complicated and has an impact or risk to the SISPQ, then the documentation level will also be complex.
The solution is to detour and filter out the no-risk changes from the comprehensive QCC system and manage (not control) them in a new system called ECM (Engineering Change Management). All aspects of the proposed non-GMP change will be managed in a simplified process, saving time and resources.
The ECM process could be designed to be paperless and flexible enough to implement such changes with minimal time, effort, and documentation. The ECM can be designed to involve all SMEs (Subject Matter Experts), including the Quality unit. Whenever there is a change with potential quality impact, this provides traceability and establishes accountability of all business functions involved in an ECM change.
The Engineering or Operations Unit usually manages the ECM process, unlike the Quality Change Control process, which is usually managed by the Quality unit to ensure the validated state is maintained. There will be no re-validation or re-qualification requirement due to an ECM change. ECM should not impact the validated state.
Establishing an ECM process fits perfectly well within the RBA (Risk-Based Approach). RBA focuses on what affects the product quality and identifies the CQAs (Critical Quality Attributes) and controls and qualifies these CQAs, thus eliminating any potential risk to the product quality. The bottom line for the RBA is to provide support rather than enforcement. Adopting the RBA would also secure business longevity.
This ECM approach could minimize the disruption to services that do not need to go through the QCC/ Quality unit for assessment and approval. Also, it will improve the documentation practices, making the Quality Change Control Process used exclusively for SISPQ-related changes.
It is vital to have some essential pre-requisites to establish an ECM program:
- Upper Management and the IT team support
- Quality unit involvement
- Solid plan and road map
- An effective, engaged, and empowered implementation team.
The plan must identify the scenarios or types of changes that would require ECM versus QCC. We could quickly identify four types/scenarios:
- The first change type is data or documentation only, in which updates are required to a non-GMP field(s) related to a GMP system, like cost or work centers
- The second type is related to supporting systems outside the GMP space, for example, change to the building security system
- The third type is related to support systems and equipment inside the GMP space like hoists, lifts, ladders, tables, etc.
- The fourth type is related to non-GMP items/components supporting/or are an integral part of a GMP equipment or systems; an example would be a transfer conveyor on a packaging line. This specific change type would require a detailed assessment and scrutiny, and the initiator of the change would be required to answer a series of questions to confirm that an ECM Form can be issued; here are examples of such questions:
- Is the component’s data documented and tested within the IQOQ?
- Does the component have a direct impact on product quality?
- Will failure of the component impact quality?
- Is the component’s data recorded in the batch record, lot release data, or other GMP-related documentation?
- Does the component have direct contact with the product?
- Does the component control SISPQ critical process elements or parameters that may affect product quality?
- Is there an independent verification of the component’s functionality and performance by the control system?
- Is the component used to create or preserve a SISPQ critical status of the system?
- Does the component require calibration?
Successful completion/answering of the above change Specific questions will allow the initiator of the change to proceed to the next stage.
Approach for ECM Scenarios 1, 2, 3 and 4
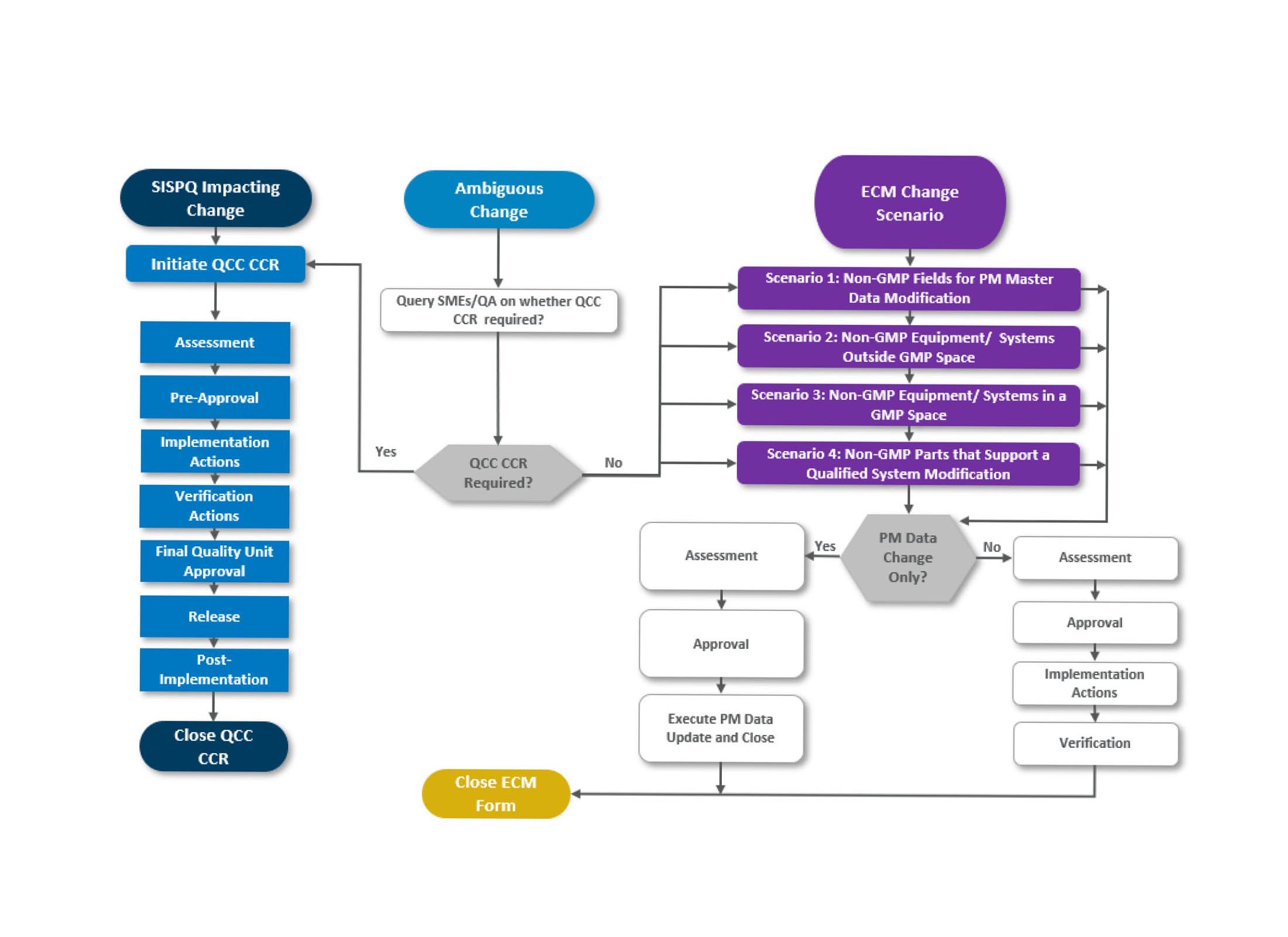
If one of the answers is not as expected, then the ECM process cannot be used to document this change, and a QCC Change Control Record would be required.
For all the ECM scenarios, the steps should generally be the same; assessment, approval, implementation, verification, and closure of the ECM form.
If the change is to data only, the steps could be less; document change and data deliverable on the ECM form, obtain approval from stakeholders, execute, and verify the change.
There are cases for a specific situation where the change is vague; thus, the decision about which process to use will be uncertain. It is recommended to default changes to the QCC process for such cases. Remember that you will not be penalized for doing more than you should, but you will be penalized if you are not doing enough.
REFERENCES
Baseline Guide Vol 5: Commissioning & Qualification 2nd Edition

ABOUT THE AUTHOR
Adnan is a Professional Engineer who has worked in the pharmaceutical and food industries for over 25 years. The following are some examples of areas of specialization:
- Change Control and Change Management systems
- Qualification of Equipment and Facilities
- Controlled Space Environmental Mapping
Adnan also has a wealth of practical expertise that enables him to influence and interact with cross-functional teams. He is a collaborative leader who motivates team members through mentoring, open dialogue, information sharing, and well-defined goals.
He is adept at making choices and solving problems. He is concurrently supporting several sites. Adnan supports the Risk Based Approach because it enables him to create and stablish systems that are effective in achieving business objectives while also upholding compliance in all associated areas, such as Equipment & Facilities Qualification.
Disclaimer: This article is based upon my personal experience and view of the ECM (Engineering Change Management) system and related ISPE Guides and does not necessarily represent the opinions of any entity with which I have been, am now, or will be affiliated.